Friction between surfaces occurs at protrusions where the surfaces contact. These protrusions are often modeled as spherical bumps on the surface, and for this reason, the prototypical model of friction is a sphere sliding on a flat surface. Here, we use large simulations to find and explain new scaling of the friction force with asperity curvature [1].
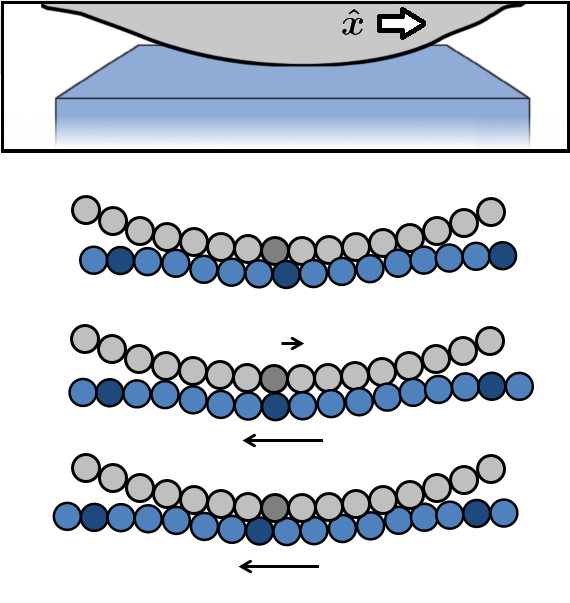
On the atomic scale, the surfaces are composed of either crystalline or disordered material, and the materials may also have loose molecules adsorbed to their surfaces. In the simplest case, the two surfaces are crystalline and clean. Furthermore, it is simplest if the crystalline surfaces are aligned (ie commensurate) and if the surfaces do not stick together (ie are non-adhesive). The other situations are more complicated, though actually more common in every-day situations, and we analyze them in other work [2]. Here, we describe the friction in the simplest, cleanest case.
Most models of friction predict a friction coefficient, 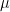
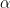


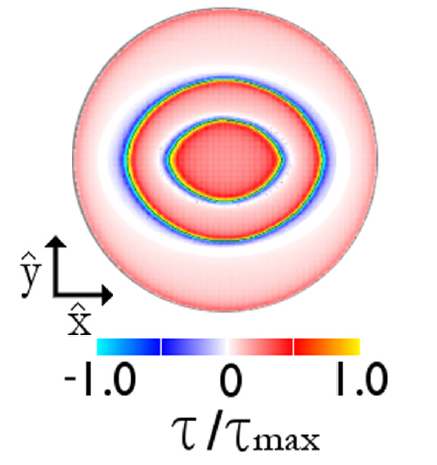
The effect of atomic geometry for this system had been considered before [4,5,6]. However, those previous studies used interactions characteristic of adhesive surfaces (tested in [2]), and did not simulate with atomic geometry. In our large-scale simulations of non-adhesive crystalline sphere sliding, we found unexpected behavior.
Fundamentally, to understand static friction, one must find the ways in which energy barriers arise in the system that oppose sliding. To understand kinetic friction, one must find the ways in which energy leaves the system. The energy is determined by the small scale atomic geometry at the surface and the long-range elastic interactions of the two materials.
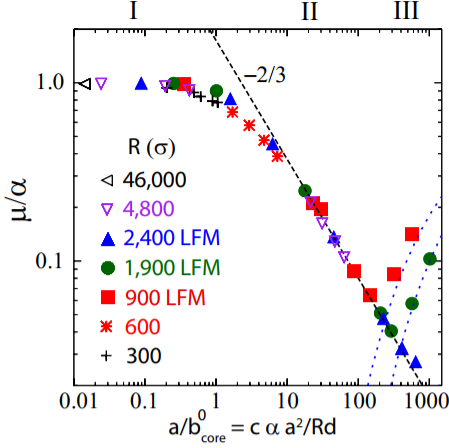
For the sphere sliding on a flat surface, elastic interactions distort the material during sliding in different ways depending on the amount of area in contact, leading to different values of the static friction coefficient. Under a sphere, the contact area is a circle, and 

In regime I, the only elastic distortion comes from the sphere dragging the elastic substrate along the direction of motion. The stress, 
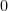

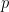

In regime II, stress builds primarily at the edges of the contact until a lattice dislocation develops and glides through the contact. The nucleation instability undercuts the ability of the friction to raise to 
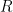
This surprising result of non-monotonic friction in the simplest manifestation of the sliding asperity model shows that friction still hides many behaviors waiting to be unraveled.
[1] Sharp, Tristan A. et al. “Scale-and load-dependent friction in commensurate sphere-on-flat contacts.” Physical Review B 96.15 (2017): 155436. https://journals.aps.org/prb/abstract/10.1103/PhysRevB.96.155436
[2] Sharp, Tristan A. et al. “Elasticity limits structural superlubricity in large contacts.” Physical Review B 93 (2016):121402(R) https://journals.aps.org/prb/abstract/10.1103/PhysRevB.93.121402
[3] Johnson, Kenneth. Contact mechanics. Cambridge university press, 1987.
Others’ work that helped build up this picture:
[4] Juan A. Hurtado and Kyung–Suk Kim. “Scale effects in friction of single–asperity contacts. I. From concurrent slip to single–dislocation–assisted slip.” Proceedings of the Royal Society A (1999): 455 1989 http://rspa.royalsocietypublishing.org/content/455/1989/3363.short
[5] Juan A. Hurtado and Kyung–Suk Kim. “Scale effects in friction of single–asperity contacts. II. Multiple–dislocation–cooperated slip” Proceedings of the Royal Society A (1999): 455 1989 http://rspa.royalsocietypublishing.org/content/455/1989/3363.short
[6] Gao, Yanfei. “A Peierls perspective on mechanisms of atomicfriction.” Journal of the Mechanics and Physics of Solids 58.12 (2010): 2023-2032. http://www.sciencedirect.com/science/article/pii/S0022509610001900
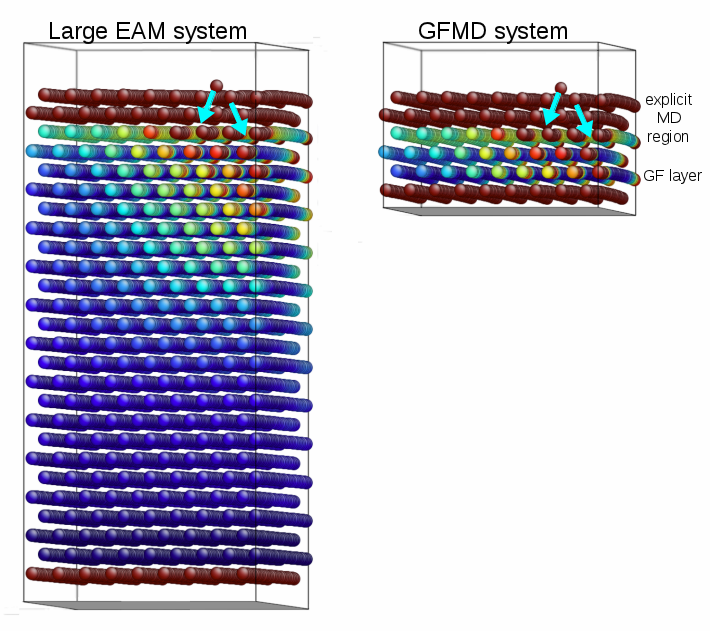
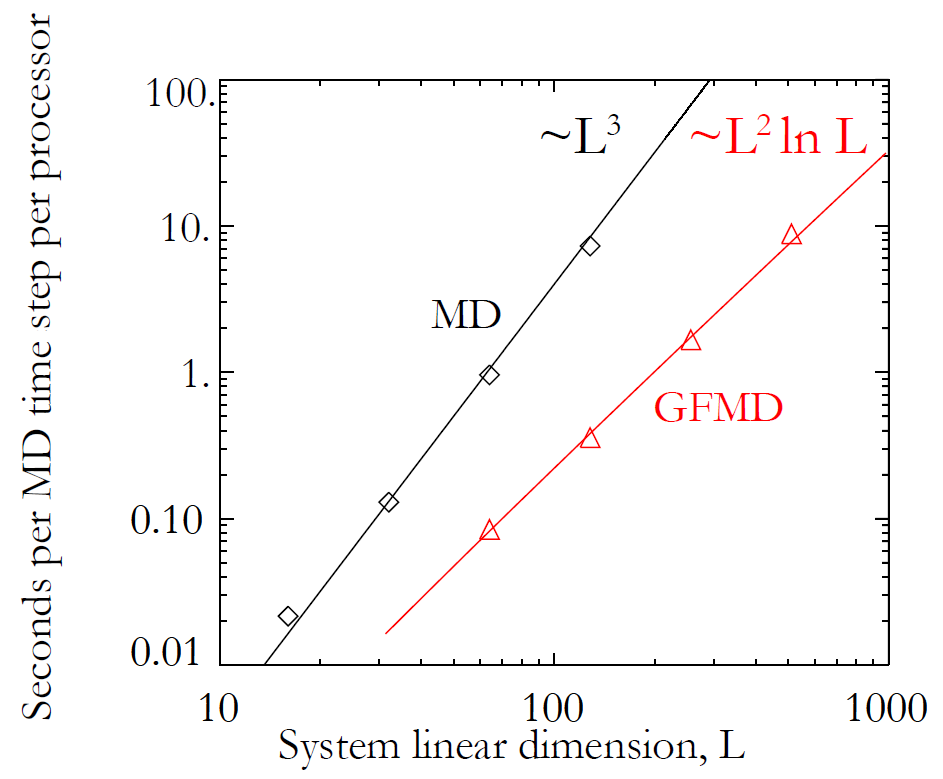
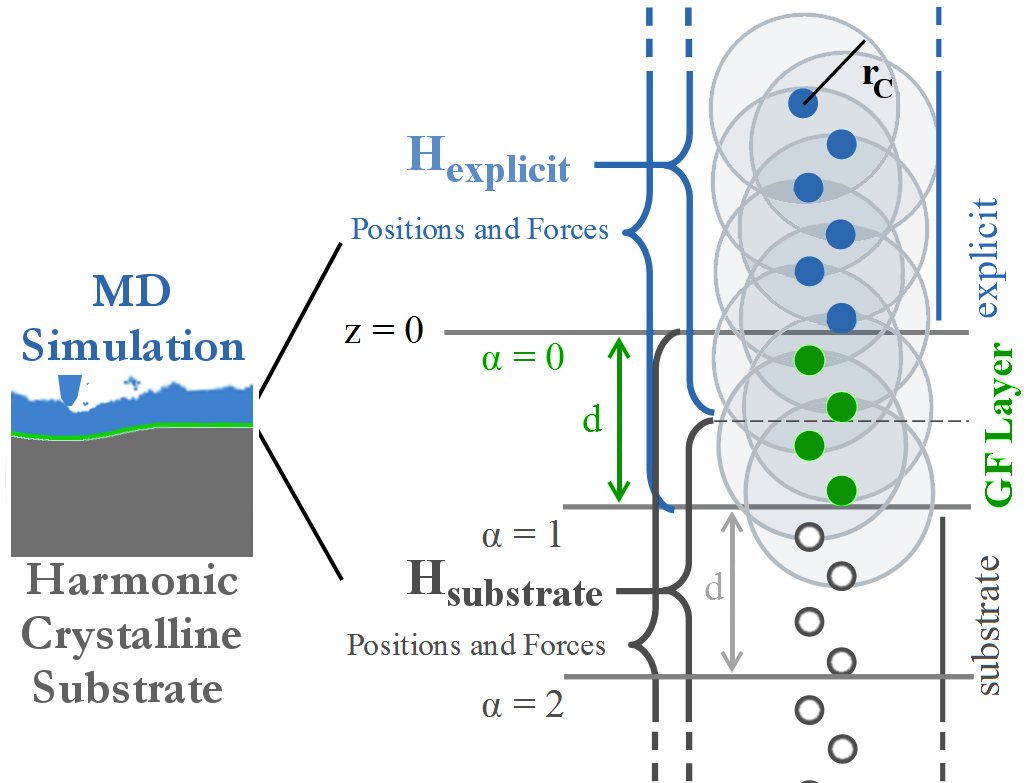
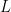 as
as  rather than
rather than  . Furthermore, the system is comprised of far fewer atoms, and so energy minimization requires fewer time steps.
. Furthermore, the system is comprised of far fewer atoms, and so energy minimization requires fewer time steps.